摘要

关键词
- 药物发现 (Drug discovery)
- 计算方法 (Computational approaches)
- 虚拟筛选 (Virtual screening)
- 人工智能 (Artificial intelligence, AI)
- 深度学习 (Deep learning, DL)
- 分子对接 (Molecular docking)
- 结构优化 (Structure optimization)
研究背景
药物发现和开发过程通常耗时且昂贵,平均需要约15年和20亿美元才能开发出一种小分子药物。计算机辅助药物设计自1970年代以来逐渐被应用,但近年来,由于数据和计算资源的爆炸性增长,相关技术得到了显著发展。虚拟筛选、深度学习和云计算的结合,为发现新颖、高效、选择性强的药物分子提供了前所未有的可能性。本文综述了当前计算技术在药物发现中的作用,重点介绍其在预筛选和配体优化中的优势,同时展望其未来潜力。
关键点
- 计算技术在药物发现中的历史与发展趋势
- 基于结构的虚拟筛选和深度学习模型的结合
- 超大规模化学空间的构建与探索
- 结合实验验证的计算方法优化
- 当前技术的局限性与未来展望
重要进展
基于结构的虚拟筛选:结构解析技术(如晶体学和冷冻电镜)的进步使得超过$90\%$的蛋白质家族拥有高分辨率3D结构模板,这为基于结构的配体筛选提供了基础。相关研究展示了虚拟筛选在快速发现高效配体中的潜力。
深度学习驱动的药物设计:结合深度学习的预测模型使得大规模化学空间的筛选更加高效,特别是在缺乏结构数据的情况下,这些模型可以预测配体的药代动力学和吸收分布代谢等特性。
化学空间的扩展:通过模块化合成技术和虚拟化学库的构建,可探索超过$10^{15}$的化学空间。这些库极大地扩展了潜在药物分子的多样性,并提高了筛选效率。
计算资源的支持:云计算和图形处理单元(GPU)的普及降低了超大规模筛选的计算成本,使虚拟筛选成为一种经济可行的药物开发策略。
结合实验的混合方法:实验验证与计算工具的结合显著提高了发现高效能配体的成功率。例如,基于计算的筛选发现了SARS-CoV-2主蛋白酶的强效抑制剂,并通过实验优化达到了纳摩尔级的效力。
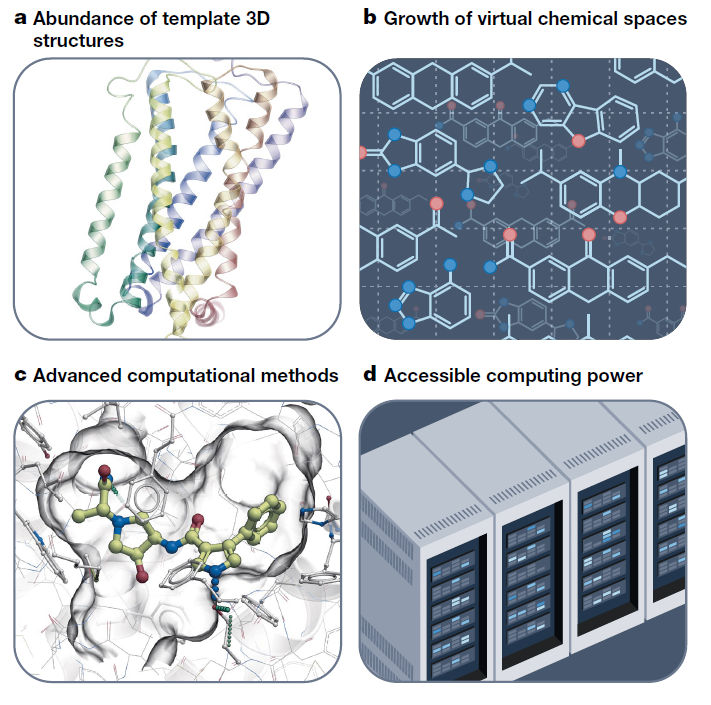
(a)蛋白质数据银行(PDB)中超过20万个蛋白质结构,以及私有数据库,已覆盖90%以上的蛋白质家族。这些结构通过高分辨率X射线晶体学和近年来的冷冻电子显微镜(cryo-EM)获得,通常处于不同的功能状态,其余的空缺也通过同源建模或AlphaFold2模型填补。(b)可用于筛选和快速合成的化学空间从2015年约10\(^7\)种现成化合物增长到2022年超过3 × 10\(^10\)种按需化合物,并可快速扩展到超过10\(^15\)种多样化且新颖的化合物。 (c)VLS的计算方法包括快速柔性对接、模块化基于片段的算法、深度学习(DL)模型和混合方法的进展。 (d)计算工具得益于经济实惠的云计算、GPU加速以及专用芯片的快速发展提供支持。
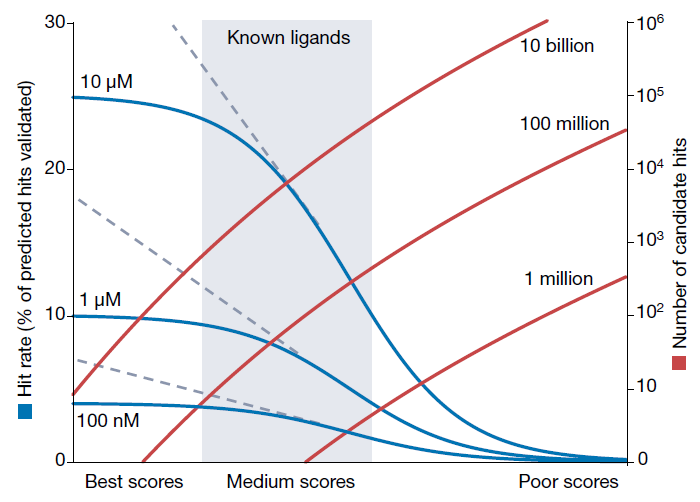
红色曲线(对数刻度)展示了在筛选库规模为100亿、1亿和100万化合物时,结合分数优于X的筛选命中分布,这些数据来源于以往的VLS和V-SYNTHES筛选研究。蓝色曲线则说明了实验命中率与预测对接分数之间的大致依赖关系,分别对应10 µM、1 µM和100 nM的阈值。尽管这种分析因靶标不同而有所变化(属于半定量性质),但其结果表明,筛选超过1亿个化合物可以突破小型化学库的局限,将命中分布的尾部向更高结合分数和更高命中率方向延伸,并能识别出更多高亲和力的实验命中物。 此外,还有两个重要因素支持将筛选库扩展到100亿及以上: (1) 候选命中物(用于合成和实验验证)通常是通过对几千个高分化合物进行靶标相关的后处理而选出的,后处理会优先考虑分子的新颖性、多样性、药物相似性,以及与特定受体残基的相互作用。因此,识别出更多高分化合物能够优化整体选择。 (2) 命中率曲线在最佳分数处的饱和并非普遍规律,而是由于筛选中使用的快速评分函数精度有限所致。在后处理步骤中,采用更精确的对接或评分方法(如柔性对接、量子力学方法或自由能扰动方法)能够将结合分数与亲和力之间的相关性向左延伸(灰色虚线),从而为超大规模化学空间带来更多高亲和力的命中物。
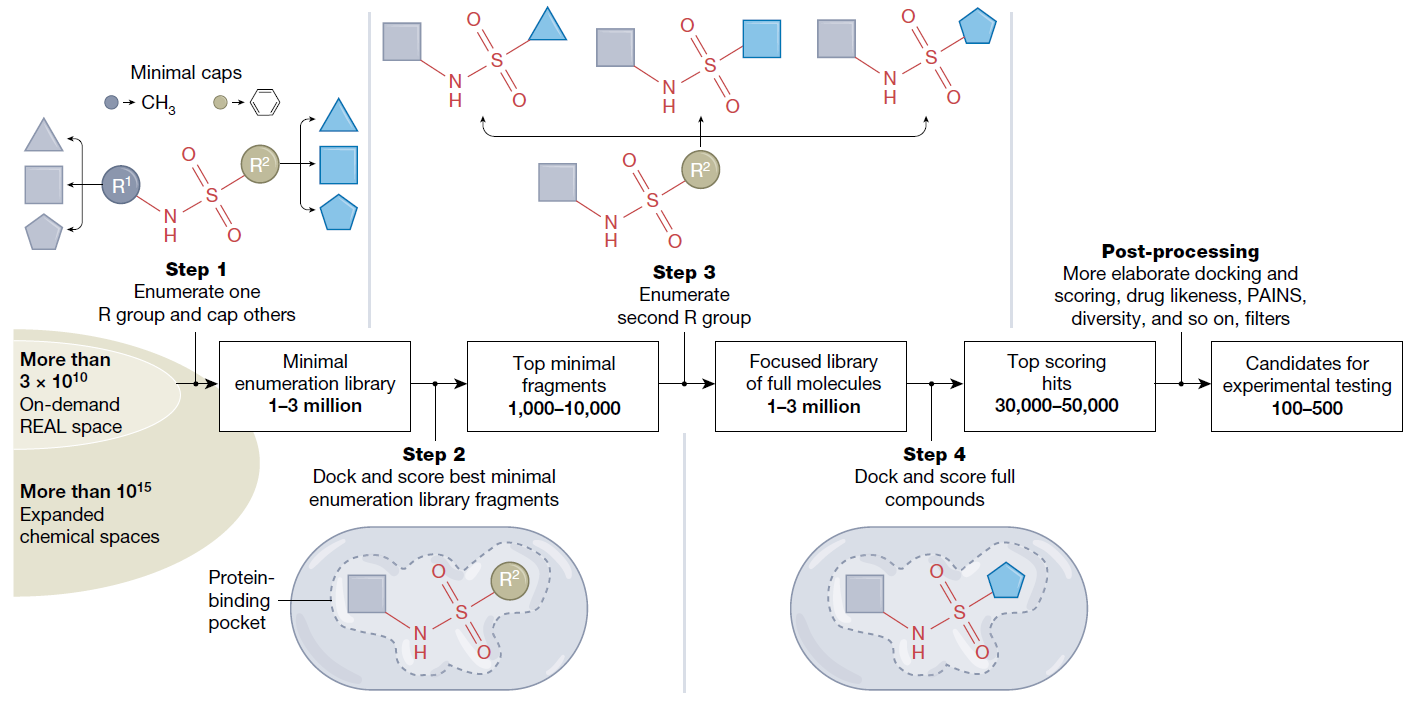
V-SYNTHES算法的概述,该算法能够在REAL Space中对超过310亿个化合物或更大的化学空间进行高效筛选,同时仅对少量分子的枚举和对接进行操作。此处以基于磺酰胺骨架的两组分反应为例,使用R1和R2合成子说明了该算法的工作原理。通过反复迭代步骤3和步骤4,该算法可应用于数百种优化的两组分、三组分或多组分反应,直至获得最佳匹配靶标结合位点的完全枚举分子。PAINS:泛筛干扰化合物(pan assay interference compounds)。

标准的高通量筛选(HTS)加定制合成驱动的发现流程与基于计算的发现流程的示意比较。后者依托于易于获取的按需或生成式虚拟化学空间,以及结构基础和人工智能驱动的计算工具,从而简化了药物发现过程中每一步的流程。
结论与展望
计算驱动的药物发现正在改变传统药物开发模式,其结合深度学习与超大规模化学库筛选的方法提供了更高效的解决方案。尽管当前技术仍面临着算法精确性和实验验证的挑战,但随着计算能力和模型精度的提高,未来有望显著降低药物开发的时间和成本,为精准医疗和个性化药物开发开辟新道路。
论文直达
原文标题:Computational approaches streamlining drug discovery
Nature 2023, 616, 673–683
点击以下链接阅读原文: