摘要

关键词
- 蛋白质折叠 (Protein Folding)
- 神经网络 (Neural Network)
- 结构生物学 (Structural Biology)
- AlphaFold (AlphaFold)
- 深度学习 (Deep Learning)
- 蛋白质结构预测 (Protein Structure Prediction)
研究背景
蛋白质结构是理解其功能的关键。然而,通过实验手段解析蛋白质结构的过程非常耗时,目前已解析的蛋白质仅占已知序列的一小部分。蛋白质折叠问题,即从氨基酸序列预测其三维结构,是生物学中的长期挑战。传统方法基于物理模拟或进化信息,通常难以实现原子级精度,特别是当缺乏同源结构时。AlphaFold通过深度学习技术,引入了新的网络架构和训练策略,融合了多序列比对和蛋白质几何约束,在CASP14评估中表现出色,为大规模结构生物信息学应用铺平了道路。
创新点
- 开发了全新的深度学习架构AlphaFold,实现了无同源结构情况下的高精度蛋白质结构预测。
- 提出Evoformer模块与结构模块的创新设计,融合了进化与几何信息。
- 在CASP14评估中表现出接近实验精度的预测能力,显著超越传统方法。
- 首次引入pLDDT评分作为预测可靠性的指标,为结果的可信度评估提供依据。
研究内容
本研究基于深度学习方法开发了AlphaFold,通过整合蛋白质的进化多序列比对和几何约束,进行蛋白质结构的高精度预测。该网络分为Evoformer和结构模块两部分:Evoformer模块通过注意力机制对多序列比对和残基对关系进行信息处理,形成蛋白质的几何假设;结构模块进一步优化预测结果,通过迭代的方式生成最终的三维结构。研究中,AlphaFold在CASP14评估中展现了远超其他方法的性能,预测结果的中位误差仅为0.96 Å。同时,通过结合预测可靠性指标pLDDT,AlphaFold提供了对预测结果的置信度评估。这种方法不仅提高了无同源结构蛋白质预测的精度,还在计算效率上表现出色,为蛋白质组学和结构生物学研究提供了重要工具。
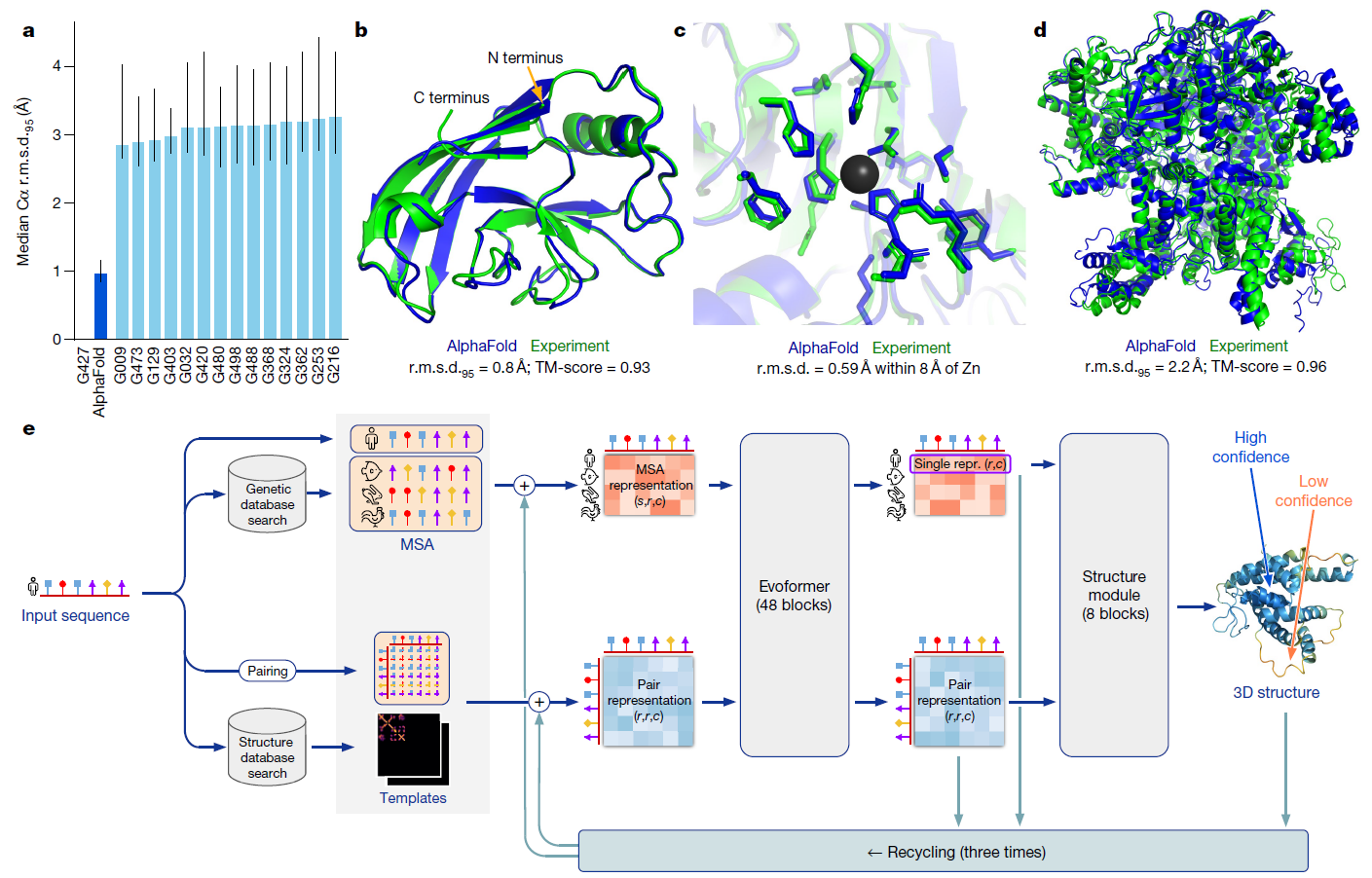
(a) AlphaFold 在 CASP14 数据集上的表现(n=87 个蛋白质结构域)相对于前 15 个条目(总计 146 个条目)的对比,组号对应 CASP 分配给参赛者的编号。数据为中位数和中位数的 95% 置信区间,由 10,000 次引导样本估计。
(b) 我们对 CASP14 目标 T1049(PDB 6Y4F,蓝色)的预测与真实(实验)结构(绿色)对比。晶体结构中 C 端的四个残基为 B 因素异常值,未显示。
(c) CASP14 目标 T1056(PDB 6YJ1)。锌结合位点的良好预测示例(尽管 AlphaFold 并未显式预测锌离子,其侧链仍然准确)。
(d) CASP 目标 T1044(PDB 6VR4)—一个由 2,180 个残基组成的单链—在 CASP 后使用 AlphaFold 无需干预预测,具有正确的结构域打包。
(e) 模型架构。箭头显示了本文中描述的各种组件之间的信息流。数组形状用括号表示,其中 \(s\) 表示序列数(主文本中的 \(N_{\text{seq}}\)),\(r\) 表示残基数(主文本中的 \(N_{\text{res}}\)),\(c\) 表示通道数。
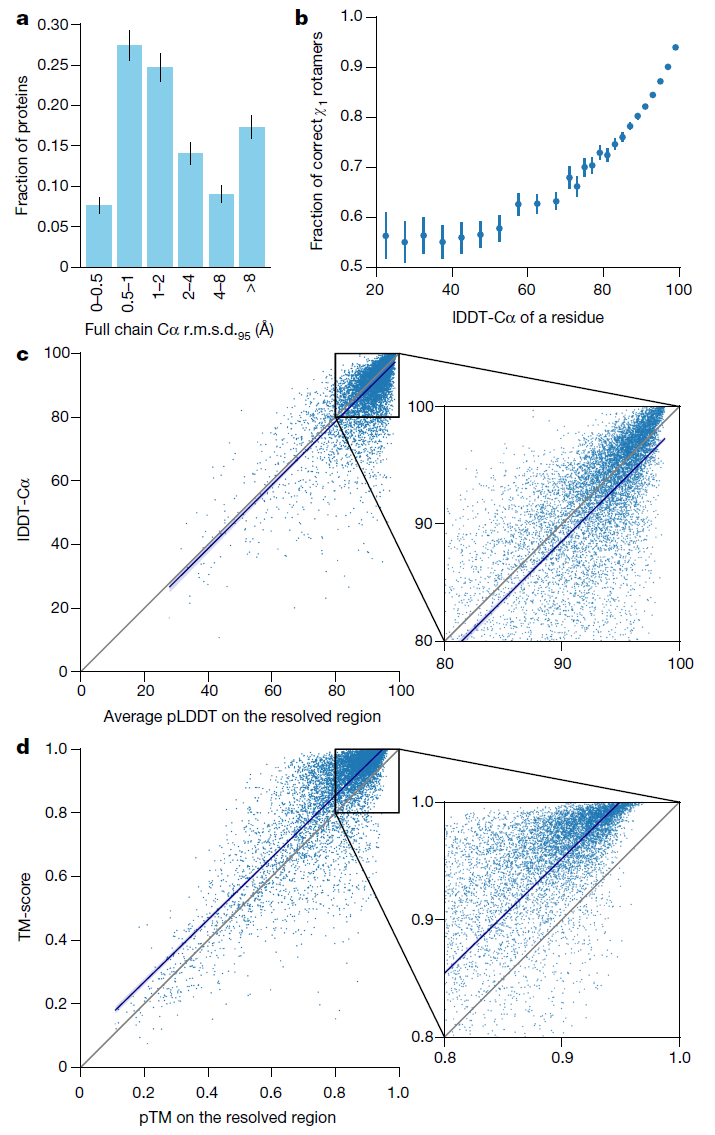
(a) 完整链(95% 覆盖的 \(C_{\alpha}\) 均方根偏差 (r.m.s.d.))的骨架 r.m.s.d. 直方图。误差条为 95% 置信区间(泊松分布)。此数据集排除了与训练集中模板(通过 hmmsearch 识别)具有超过 40% 序列身份且覆盖链的超过 1% 的蛋白质(n=3,144 个蛋白质链)。整体中位数为 1.46 Å(95% 置信区间 = 1.40–1.56 Å)。请注意,对于某些具有不确定打包或打包错误的链,预期会有较高的 r.m.s.d。
(b) 骨架精度与侧链精度之间的相关性。过滤为任意观察到侧链且分辨率优于 2.5 Å 的结构(n=5,317 个蛋白质链);进一步将侧链过滤为 \(B\) 因素 <30 Ų。若预测的扭转角在 40° 以内,则分类为正确的旋转异构体。每个点聚合一范围的 \(l_{\text{DDT}-C_{\alpha}}\),bin 尺寸为 70 \(l_{\text{DDT}-C_{\alpha}}\) 以上为 2 单位,其他情况为 5 单位。点对应平均精度;误差条为每残基基础上平均值的 95% 置信区间(学生 t 检验)。
(c) 置信分数与链的真实精度的比较。最小二乘线性拟合 \(l_{\text{DDT}-C_{\alpha}} = 0.997 \times p_{\text{LDDT}} - 1.17\)(Pearson 的 \(r = 0.76\))。\(n = 10,795\) 条蛋白链。线性拟合的阴影区域表示从 10,000 个引导样本中估算的 95% 置信区间。
(d) \(p_{\text{TM}}\) 与完整链 TM-score 之间的相关性。最小二乘线性拟合 \(TM-\text{score} = 0.98 \times p_{\text{TM}} + 0.07\)(Pearson 的 \(r = 0.85\))。\(n = 10,795\) 条蛋白质链。线性拟合的阴影区域表示从 10,000 个引导样本中估算的 95% 置信区间。
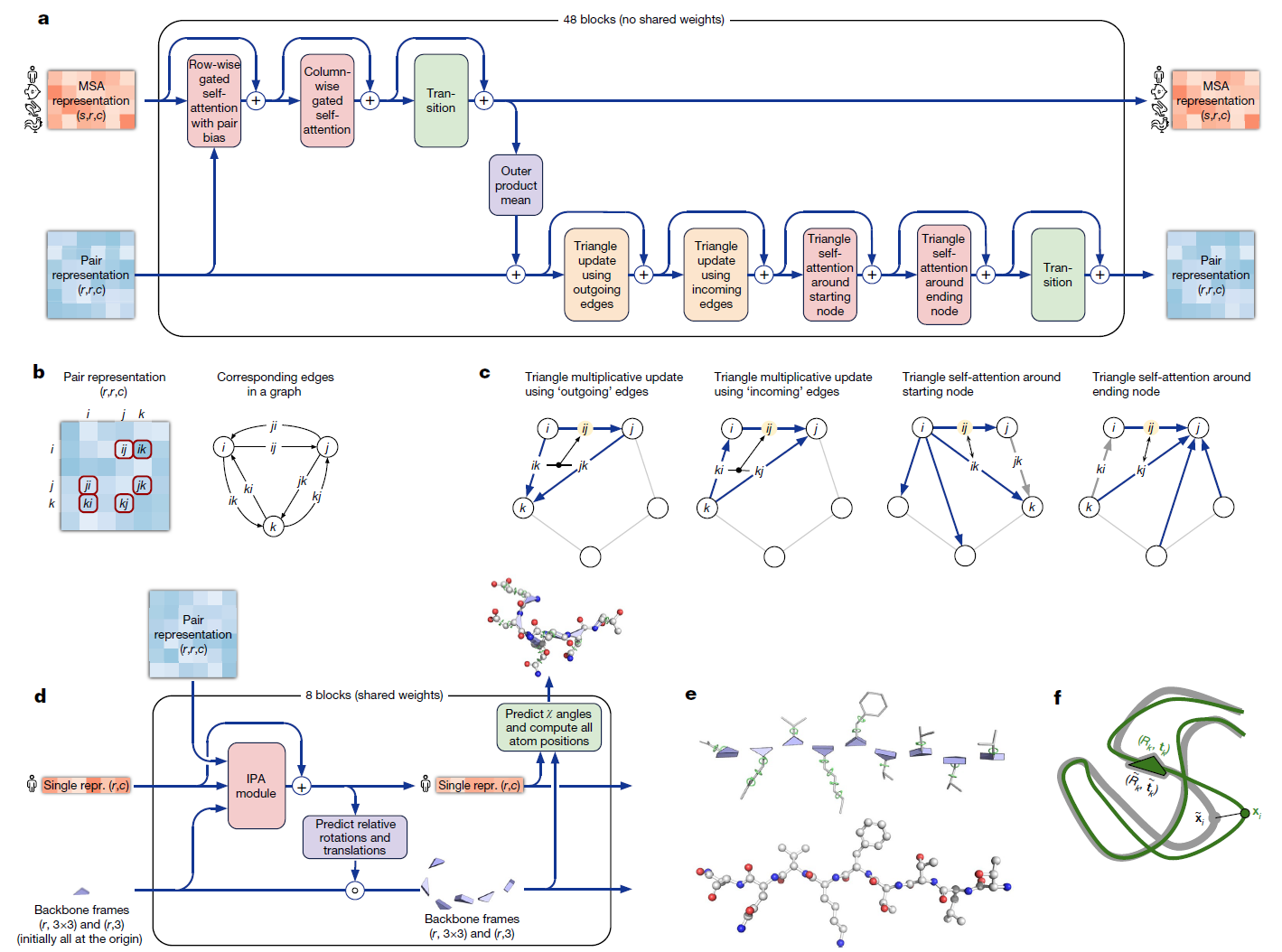
(a) Evoformer 模块。箭头显示了信息流。数组的形状用括号表示。
(b) 作为图中有向边解释的对表示。
(c) 三角乘法更新和三角自注意力。圆圈表示残基。对表示中的条目显示为有向边,并在每个图中显示正在更新的边 ij。
(d) 结构模块,包括不变点注意力(IPA)模块。单个表示是 MSA 表示的第一行的副本。
(e) 残基气体:每个残基的表示为骨架的一个自由浮动刚体(蓝色三角形)和侧链的 χ 角(绿色圆圈)。下方显示对应的原子结构。
(f) 框架对齐点误差(FAPE)。绿色,预测结构;灰色,真实结构;(Rk, tk),框架;xi,原子位置。
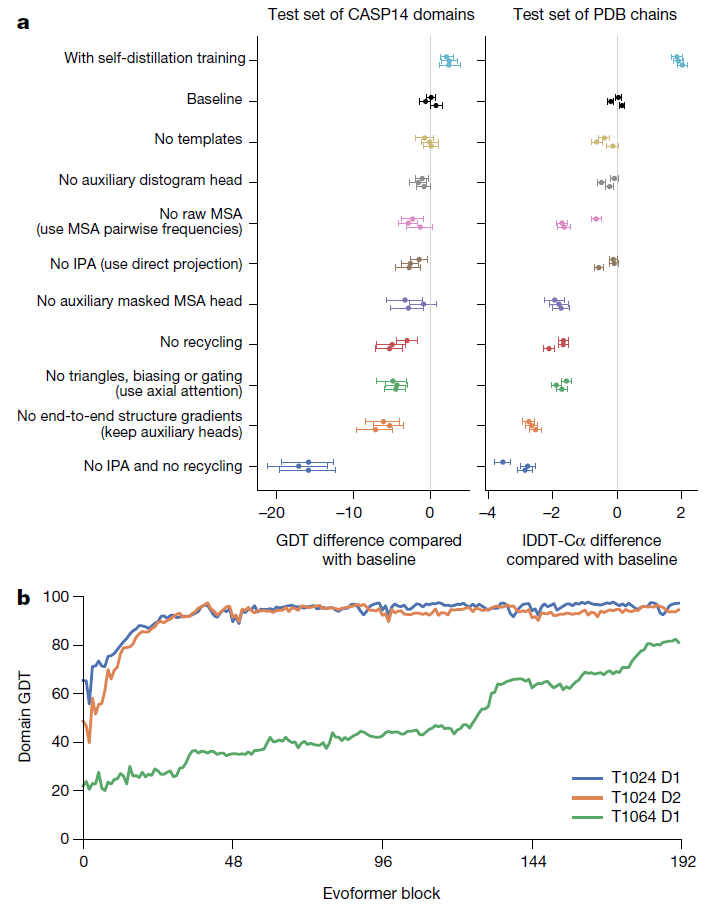
(a) 消融实验。对 CASP14 和最近 PDB 集合中的链进行不同网络设计的改动分析。分数为相对于基线网络的变化。
(b) 在不同 Evoformer 模块层数上的 GDT-C 轨迹,对 SARS-CoV-2 ORF8 和 CASP14 的其他目标的预测路径进行比较。
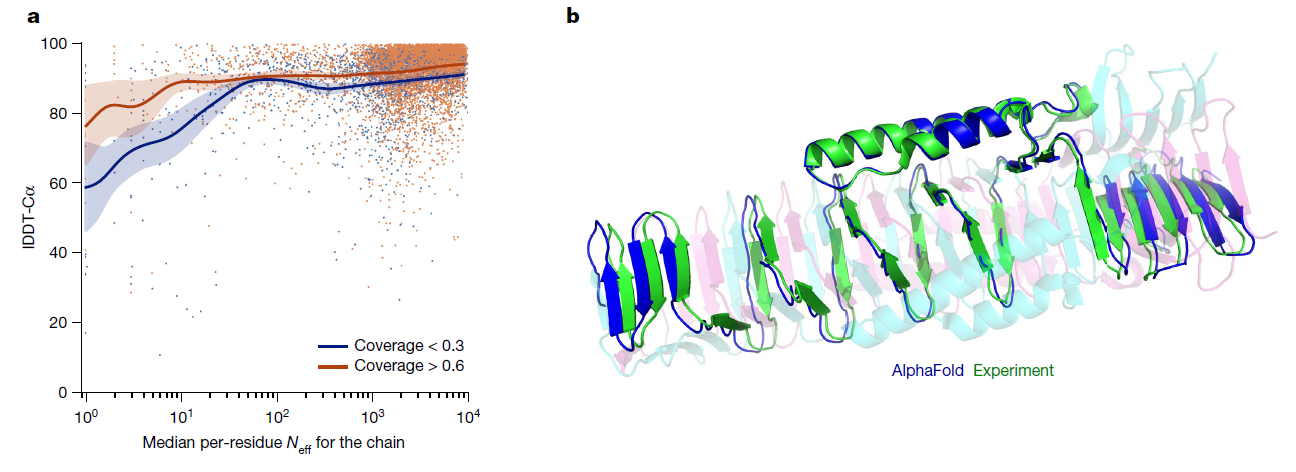
(a) MSA 深度对骨架精度的影响,lDDT-C\alpha 曲线与覆盖情况比较。
(b) 一个同源三聚体的交织(PDB 6SK0)结构正确预测,无需输入化学计量比且仅具有弱模板。
结论与展望
AlphaFold的开发标志着蛋白质结构预测领域的重大突破,其在CASP14中的表现验证了该方法的可靠性与准确性。未来,该方法有望应用于复杂蛋白质-蛋白质相互作用预测和完整蛋白质复合物的建模。同时,随着数据库的扩展与算法的改进,AlphaFold将为生物医学和制药领域提供更广泛的应用可能性。
论文直达
原文标题:Highly accurate protein structure prediction with AlphaFold
Nature 2021, 596, 583–589.
点击以下链接阅读原文: